血管平滑肌细胞(VSMCs)位于动脉血管中膜,维持动脉结构完整性和血管张力。已在 VSMCs 中鉴定出一系列表型。VSMCs 在生理条件下处于静止状态并呈现收缩表型。当受到有害刺激时,VSMCs 会失去收缩特性并转化为合成表型,这对动脉粥样硬化斑块的形成和进展至关重要。研究表明,delta-样配体4-(Dll4-)激活的 Notch 信号通路在 VSMC 收缩合成表型转换中是必不可少的。据报道,DLL4 阻断有效减轻包括动脉粥样硬化在内的几种代谢紊乱。动脉粥样硬化(AS)斑块的形成、进展和演变与巨噬细胞高度相关,巨噬细胞响应不同的刺激而有不同的极化。一般来说,巨噬细胞主要可分为 M1 型即经典活化的巨噬细胞,和 M2 型即替代性活化的巨噬细胞。由于它们的促炎特性,M1 巨噬细胞被认为有助于动脉粥样硬化病变的生长和斑块的不稳定性。根据最近的一项研究,静止的巨噬细胞(M0)到 M1 的极化可以由各种代谢物驱动,例如晚期糖基化终产物(AGEs)。AGEs 是 2 型糖尿病(T2DM)的一组代表性代谢物,与 AS 高度相关。通过与 AGEs 受体(RAGE)相互作用,AGEs 引发各种病理结果。最近的研究表明,AGEs 和 RAGE 之间的相互作用激活了 toll 样受体4(TLR4)信号通路,从而促进了巨噬细胞 M0 到 M1 的极化。初步研究证实,AGE 诱导的 M1 极化巨噬细胞具有高表达 Dll4 的特点。因此,假设 AGEs 在 M0 至 M1 极化期间赋予巨噬细胞高表达 Dll4。这些巨噬细胞将通过激活 Notch 信号进一步促进 VSMCs 进行从收缩到合成的表型转换。在陕西省人民医院心血管内科的一项研究中,T2DM AS 动物被给予 TLR4 的特异性抑制剂 TAK-242,观察抗 AS 效应。M0 巨噬细胞暴露于外源性 AGEs,然后与原代 VSMCs 共培养。巨噬细胞 TLR4 信号通路被特异性小干扰 RNA(siRNAs)抑制。这项研究的结果将增加对糖尿病相关AS机制的更多了解,并为未来潜在的基因靶向治疗提供更多线索。抑制 TLR4 抑制以高血清 AGE 浓度为特征的 AS DM 动物动脉粥样硬化病变中动脉狭窄、M1 巨噬细胞浸润和 VSMC 表型转换与 AS 对照相比,AS DM 小鼠的血清空腹血糖(FBG)和 AGE 浓度显著升高(图1 a)。在从 AS DM 动物取样的主动脉根部中发现了显著增加的动脉狭窄、诱导型一氧化氮合酶(iNOS)、RAGE、TLR4 表达和降低的肌球蛋白重链11(MYH11)表达(图1 b-d)。TLR4 抑制剂 TAK-242 的给药未能影响 AS DM 小鼠的血清 FBG 和 AGE 浓度(图1 a)。在 TAK-242 给药的 AS DM 小鼠的 AS 病变中发现动脉狭窄、iNOS 和 TLR4 表达显著降低,但 MYH11 表达增加(图1 b-d)。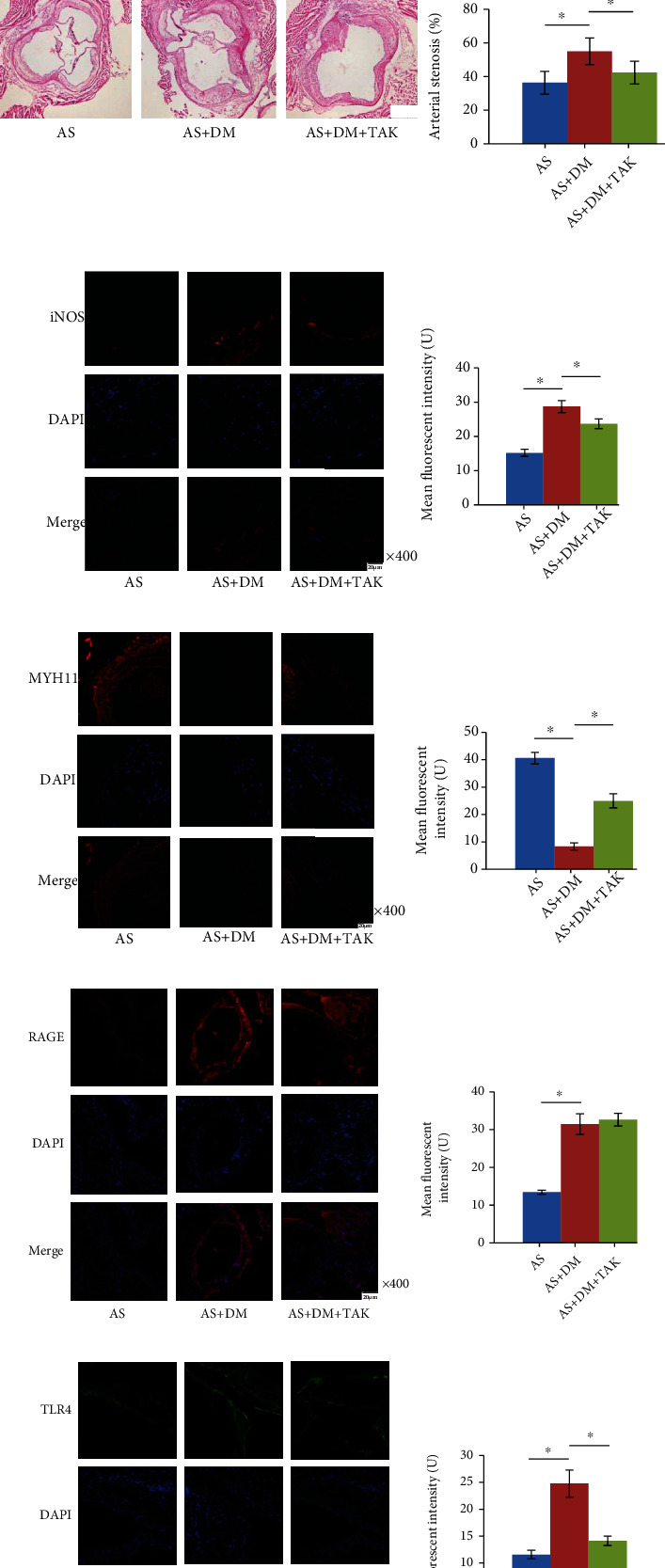 (a)表示空腹血糖(FGB)和血清 AGE 浓度。(b)主动脉根部的 H&E 染色显示动脉粥样硬化斑块。(c)捕获的 iNOS 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(d)捕获的 MYH11 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(e)捕获的 RAGE 免疫荧光染色图像。还演示了 DAPI 及其合并图像。(f)捕获的 TLR4 的免疫荧光染色图像。还演示了 DAPI 及其合并图像。AS:动脉粥样硬化小鼠;AS+DM:动脉粥样硬化糖尿病小鼠;AS+DM+TAK:用 TAK-242 处理的动脉粥样硬化糖尿病小鼠靶向沉默的 RAGE/TLR4 通路抑制 AGE-BSA 诱导的巨噬细胞 M0 至 M1 极化将原代 M0 巨噬细胞暴露于 0、100 和 200 μg/mL 的 AGE-牛血清白蛋白(BSA)48 小时。如图2 a 所示,AGE-BSA 暴露增加了 iNOS 的表达,iNOS 被认为是巨噬细胞 M1 表型的标志物, 且呈 AGE-BSA 浓度依赖性方式。同时,如图2 b、c所示,AGE-BSA 暴露以 AGE-BSA 浓度依赖性方式增加巨噬细胞中 RAGE 和 TLR4 的表达水平。暴露还以 AGE-BSA 浓度依赖性方式增加促炎细胞因子的分泌,包括 IL1β 、IL6 和 TNFα(图2 d)。在暴露于 200 μg/mL AGE-BSA 的巨噬细胞中,靶向沉默 RAGE 和 TLR4 被证明可以有效(图2 b、c)降低 iNOS 表达(图2 a)和促炎细胞因子浓度(图2 d)。
(a)表示空腹血糖(FGB)和血清 AGE 浓度。(b)主动脉根部的 H&E 染色显示动脉粥样硬化斑块。(c)捕获的 iNOS 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(d)捕获的 MYH11 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(e)捕获的 RAGE 免疫荧光染色图像。还演示了 DAPI 及其合并图像。(f)捕获的 TLR4 的免疫荧光染色图像。还演示了 DAPI 及其合并图像。AS:动脉粥样硬化小鼠;AS+DM:动脉粥样硬化糖尿病小鼠;AS+DM+TAK:用 TAK-242 处理的动脉粥样硬化糖尿病小鼠靶向沉默的 RAGE/TLR4 通路抑制 AGE-BSA 诱导的巨噬细胞 M0 至 M1 极化将原代 M0 巨噬细胞暴露于 0、100 和 200 μg/mL 的 AGE-牛血清白蛋白(BSA)48 小时。如图2 a 所示,AGE-BSA 暴露增加了 iNOS 的表达,iNOS 被认为是巨噬细胞 M1 表型的标志物, 且呈 AGE-BSA 浓度依赖性方式。同时,如图2 b、c所示,AGE-BSA 暴露以 AGE-BSA 浓度依赖性方式增加巨噬细胞中 RAGE 和 TLR4 的表达水平。暴露还以 AGE-BSA 浓度依赖性方式增加促炎细胞因子的分泌,包括 IL1β 、IL6 和 TNFα(图2 d)。在暴露于 200 μg/mL AGE-BSA 的巨噬细胞中,靶向沉默 RAGE 和 TLR4 被证明可以有效(图2 b、c)降低 iNOS 表达(图2 a)和促炎细胞因子浓度(图2 d)。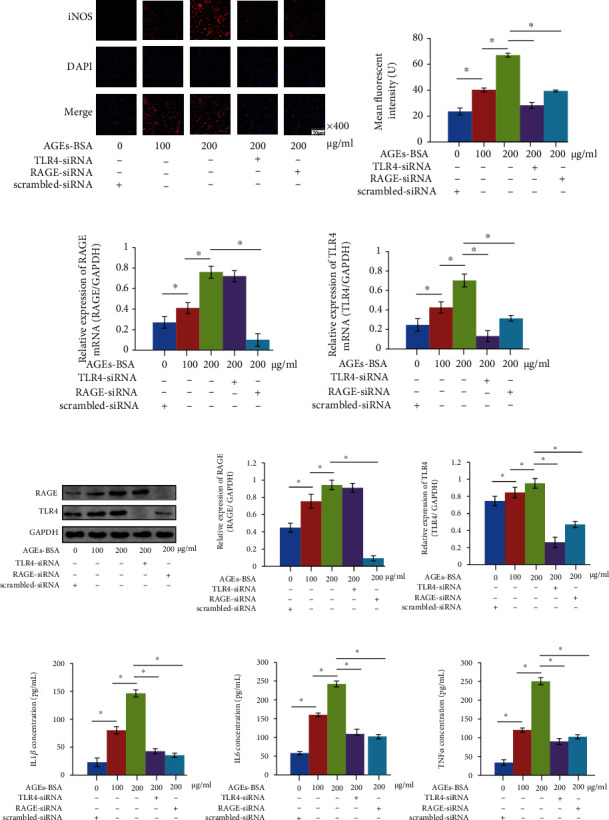 (a)捕获的 iNOS 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(b)表示巨噬细胞中RAGE和TLR4的相对mRNA表达水平。(c)免疫印迹显示巨噬细胞中 RAGE、TLR4 和 GAPDH 。右图的列表示巨噬细胞中 RAGE 和 TLR4 的相对表达水平。(d)表示巨噬细胞的细胞培养基上清液中IL1β、IL6 和 TNFα的浓度。沉默 RAGE/TLR4 通路降低 AGE-BSA 孵育的巨噬细胞中 Dll4 的表达在巨噬细胞中,AGE-BSA 暴露显著增加了细胞外调节蛋白激酶(ERK)磷酸化水平(图3 a)以及叉头框蛋白C2(FOXC2) 和 Dll4 表达水平(图3 b),且呈 AGE-BSA 浓度依赖性方式。在暴露于 200 μg/mL AGE-BSA 的巨噬细胞中,RAGE 和 TLR4 沉默显著抑制 ERK 磷酸化(图3 a)并降低 FOXC2 和 Dll4 的表达水平(图3 b)。
(a)捕获的 iNOS 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。(b)表示巨噬细胞中RAGE和TLR4的相对mRNA表达水平。(c)免疫印迹显示巨噬细胞中 RAGE、TLR4 和 GAPDH 。右图的列表示巨噬细胞中 RAGE 和 TLR4 的相对表达水平。(d)表示巨噬细胞的细胞培养基上清液中IL1β、IL6 和 TNFα的浓度。沉默 RAGE/TLR4 通路降低 AGE-BSA 孵育的巨噬细胞中 Dll4 的表达在巨噬细胞中,AGE-BSA 暴露显著增加了细胞外调节蛋白激酶(ERK)磷酸化水平(图3 a)以及叉头框蛋白C2(FOXC2) 和 Dll4 表达水平(图3 b),且呈 AGE-BSA 浓度依赖性方式。在暴露于 200 μg/mL AGE-BSA 的巨噬细胞中,RAGE 和 TLR4 沉默显著抑制 ERK 磷酸化(图3 a)并降低 FOXC2 和 Dll4 的表达水平(图3 b)。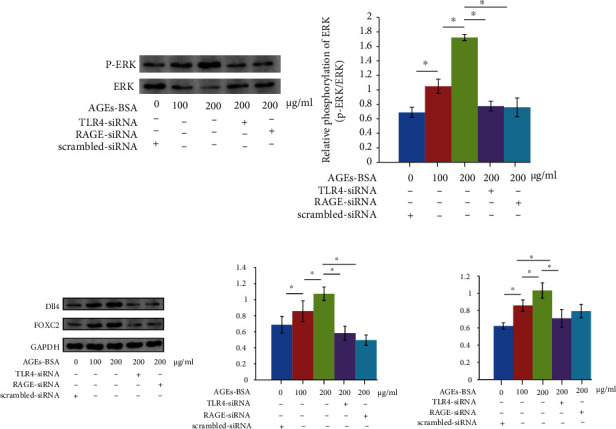 (a)ERK 和 p-ERK 的免疫印迹显示在左图中。列表示巨噬细胞中 ERK 的相对磷酸化水平。(b)FOXC2、Dll4 和 GAPDH 的免疫印迹。右图的列表示巨噬细胞中 FOXC2 和 Dll4 的相对表达水平。RAGE/TLR4 通路的沉默削弱了 AGE 暴露的巨噬细胞在接触共培养模型中诱导 VSMC 收缩-合成表型转换的能力已建立的接触式和非接触式共培养模型在图4 a。如图4 b,在非接触模型中,AGE-BSA 暴露和 AGE-BSA 未暴露的巨噬细胞均未能改变 VSMC 中的 MYH11 表达。然而,在接触模型中,AGE-BSA 暴露的巨噬细胞显著诱导 VSMC 表型转换,这可以通过 VSMCs 中 MYH11 表达的改变来证明。如图4 c 所示,在接触共培养模型中,与 RAGE 和 TLR4 沉默的 AGE-BSA 暴露的巨噬细胞共培养的 VSMCs 表现出 NICD1 和 HES 的表达水平降低,这是 Notch 信号通路激活的标志物。然而,在非接触模型中,NICD1 和 HES1 的表达水平在 VSMCs 中没有改变(图4 d)。
(a)ERK 和 p-ERK 的免疫印迹显示在左图中。列表示巨噬细胞中 ERK 的相对磷酸化水平。(b)FOXC2、Dll4 和 GAPDH 的免疫印迹。右图的列表示巨噬细胞中 FOXC2 和 Dll4 的相对表达水平。RAGE/TLR4 通路的沉默削弱了 AGE 暴露的巨噬细胞在接触共培养模型中诱导 VSMC 收缩-合成表型转换的能力已建立的接触式和非接触式共培养模型在图4 a。如图4 b,在非接触模型中,AGE-BSA 暴露和 AGE-BSA 未暴露的巨噬细胞均未能改变 VSMC 中的 MYH11 表达。然而,在接触模型中,AGE-BSA 暴露的巨噬细胞显著诱导 VSMC 表型转换,这可以通过 VSMCs 中 MYH11 表达的改变来证明。如图4 c 所示,在接触共培养模型中,与 RAGE 和 TLR4 沉默的 AGE-BSA 暴露的巨噬细胞共培养的 VSMCs 表现出 NICD1 和 HES 的表达水平降低,这是 Notch 信号通路激活的标志物。然而,在非接触模型中,NICD1 和 HES1 的表达水平在 VSMCs 中没有改变(图4 d)。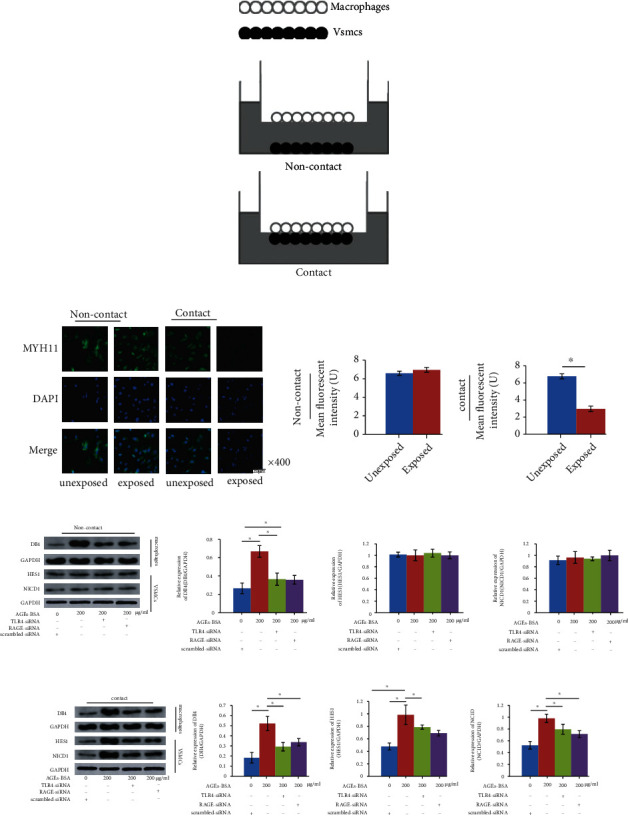 (b)捕获的 MYH11 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。右列分别表示 MYH11 在接触和非接触共培养模型中的平均荧光强度。(c)非接触共培养模型中,巨噬细胞中的 Dll4、GAPDH 和 VSMCs 中的 HES1、NICD1 和 GAPDH 的免疫印迹。列表示非接触共培养模型中巨噬细胞中的Dll4和VSMCs 中HES1 和 NICD1 的相对表达水平。(d)接触共培养模型中巨噬细胞中 Dll4、GAPDH 和 VSMCs 中 HES1、NICD1 和 GAPDH 的免疫印迹。列表示接触共培养模型中巨噬细胞中的Dll4和VSMCs 中HES1 和 NICD1 的相对表达水平。
(b)捕获的 MYH11 的免疫荧光染色图像,还演示了 DAPI 及其合并图像。右列分别表示 MYH11 在接触和非接触共培养模型中的平均荧光强度。(c)非接触共培养模型中,巨噬细胞中的 Dll4、GAPDH 和 VSMCs 中的 HES1、NICD1 和 GAPDH 的免疫印迹。列表示非接触共培养模型中巨噬细胞中的Dll4和VSMCs 中HES1 和 NICD1 的相对表达水平。(d)接触共培养模型中巨噬细胞中 Dll4、GAPDH 和 VSMCs 中 HES1、NICD1 和 GAPDH 的免疫印迹。列表示接触共培养模型中巨噬细胞中的Dll4和VSMCs 中HES1 和 NICD1 的相对表达水平。总之,该研究数据提出了一种新的分子机制来解释 DM 如何加剧 AS。DM 促进的 AGEs 诱导斑块巨噬细胞中 RAGE/TLR4 通路的激活,这些通路执行 M1 极化。在此过程中,激活的 RAGE/TLR4 信号通过 ERK/FOXC2 通路在该斑块 M1 巨噬细胞亚群中促进 Dll4 表达。AGE 处理的巨噬细胞通过在接触共培养模型而不是非接触模型中激活 Notch 通路诱导 VSMC 表型转化,从而促成 AS。抑制 TLR4 信号使M1巨噬细胞失去 Dll4 高表达表型。因此,VSMC 表型转换被抑制,糖尿病加重的 AS 被减弱。
参考文献:Xing Y, Pan S, Zhu L, Cui Q, Tang Z, Liu Z, Liu F. Advanced Glycation End Products Induce Atherosclerosis via RAGE/TLR4 Signaling Mediated-M1 Macrophage Polarization-Dependent Vascular Smooth Muscle Cell Phenotypic Conversion. Oxid Med Cell Longev. 2022 Jan 13;2022:9763377. doi: 10.1155/2022/9763377. PMID: 35069982; PMCID: PMC8776434.原文链接:https://pubmed-ncbi-nlm-nih-gov.proxy.library.carleton.ca/35069982/小编旨在分享、学习、交流生物科学等领域的研究进展。如有侵权或引文不当请联系小编修正。微信搜索公众号“Naturethink”,学习更多关于流体机械力学刺激细胞培养的知识吧!